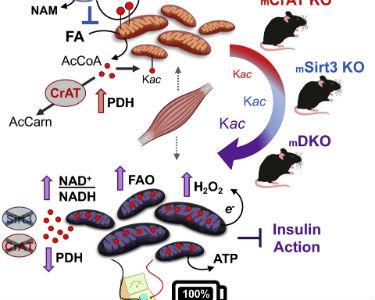
A team of researchers at DMPI led by Postdoctoral Associate, Ashley Williams, and Professor of Medicine, Deb Muoio, sought to understand why mitochondria–the primary metabolic engines in most cells–begin to fail when organisms are exposed to chronic overnutrition and obesity. One widely accepted theory suggests that surplus carbons derived from dietary fats react with oxidative machinery resident within the mitochondrial matrix, resulting in post-translational protein modifications (PTMs) known as lysine acetylation. Mounting circumstantial evidence has led to the presumption that acetyl-lysine PTMs damage mitochondrial proteins, which in turn disrupts respiratory performance, lowers lipid catabolism and compromises metabolic control.
Results of the new study challenge this theory by showing that knockout mice engineered to have deficient deacetylase activity and extreme levels of acetyl-lysine PTMs in muscle mitochondria do not show signs of mitochondrial damage or respiratory failure; but instead, have elevated propensity to oxidize fat while also developing more severe diet-induced glucose intolerance and insulin resistance. The surprising findings led the DMPI team to conclude that the process of lysine deacetylation imposes a brake on lipid catabolism, and thereby plays a role in shifting muscle fuel use from fat to glucose during transitions from fasting to feeding. According to this model, acetyl-lysine turnover, not PTM-mediated protein damage, regulates mitochondrial fuel choice and glucose homeostasis. The findings provide new insights into the connection between obesity, type 2 diabetes and mitochondrial health.